
01/基于分子对接的虚拟筛选
分子对接是一种基于靶标蛋白三维结构的药物筛选方式。
通过小分子化合物与靶标进行分子对接,综合分析得分及空间构象情况, 包括静电作用、氢键作用、疏水作用、范德华作用等性质, 可以探索配体小分子与受体生物大分子具体作用方式和结合构型, 解释化合物产生活性的原因,为合理地优化化合物结构提供指导; 通过结合亲合性打分函数,小分子化合物与靶蛋白的结合能力可以定量地得到评价, 为候选化合物的挑选提供依据,筛选潜在活性化合物,为实验提供参考。
基于分子对接的小分子化合物的虚拟筛选是目前最有潜力的药物开发工具, 由于分子对接过程对化合物结构并没有限制,基于分子对接的虚拟筛选完全有可能获得结构新颖的先导化合物。
可以有效的节约时间和费用,这对于发展创新药物无疑起到巨大的推动作用。
02/基于药效团的虚拟筛选
药效团是一种基于小分子化合物的高效药物筛选手段。
通过分析一个或多个活性分子的药效特征,推测使分子具有活性的重要药效基团特征。
药效团筛选的计算量较小,可以在分子对接前进行, 对几百万或几千万的小分子数据库进行药效团筛选只需要很短时间。
客户只需要提供1个或多个活性分子,就可以构建公共药效团进行筛选, 搜索含相同特征的小分子,并指导新活性分子的合成。
一些分子模拟软件还提供方法来预测匹配分子的生物活性,如基于药效团的QSAR模型等。
但是,基于药效团模型的虚拟筛选通常获得与构建模型所用的化合物结构性质类似的分子结构, 在发展新颖骨架的先导化合物方面具有一定的局限性。
典型案例
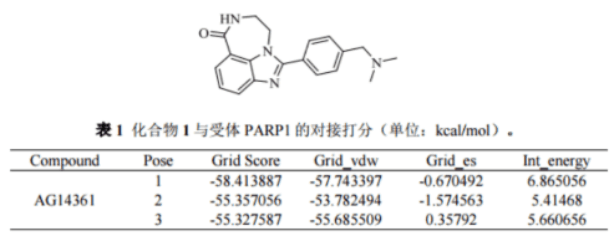
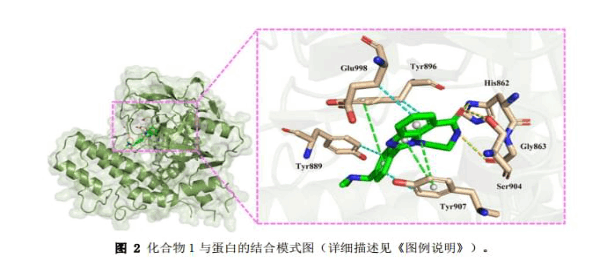
在本案例中我们采用分子对接技术研究化合物1的结构(如左图)与PARP1蛋白的结合能力, 采用 DOCK 6 .7 程序预测化合物1在PARP1中的结合模式,保留最多 20 个结合构象。
计算结果表明,结合位点均有多个对接构象,其打分情况(左表 1), 根据打分和结合模式选取第二个对接构象进行结合模式分析。
化合物1七元环上的酰胺羰基氧原子与氨基酸残基Ser904和His862形成氢键相互作用; 同时,酰胺氮原子与 Gly863 形成了 3.33 Å 的氢键相互作用。这为化合物的结合锚定了方向, 并提供了一定的静电力贡献(Grid_es = -1.574563 kcal/mol)。
苯并咪唑的两个环与Tyr90之间形成P型π-π堆积作用,芳环中心距禝分别为 4.21 Å 和 4.93 Å; 侧链苯环与Tyr896之间形成T型π-π堆积作用,芳环中心距禝为 5.47 Å。 同时,化合物还与残基Tyr889、Tyr896、Tyr907和 Glu998之间形成疏水作用, 疏水作用和π-π堆积作用为化合物提供了强大的范德华力(Grid_vdw = -53.78 kcal/mol)。 综上,化合物1与蛋白 PARP1的相互作用以 π-π 堆积和疏水作用为主,并通过氢键作用锁定结合取向(图2)。